North America


International Markets
























































North America
International Markets
Europe
Generic Medicines and Regulation: Producing the World’s Biggest Medicine Cabinet Safely and Effectively


Most people know what generic medicines are - products created to be the same as branded drugs whose patents have expired. They have the same active ingredients and clinical effects as branded medicines and are also as safe to use. What’s less well known is how these more affordable versions of branded drugs are regulated to ensure that they are safe and effective.
Teva believes that everybody should have access to high quality medicines that help manage disease, fight infection, or simply improve overall health. That’s why we’re focusing a significant portion of our research and development budget on generic medicine. In 2020, Teva invested nearly $1 billion in R&D activities and a large number of Teva medicines continue to be used to fill prescriptions around the world.

Why do generic medicines cost less than branded drugs?
“When a medicine is originally developed, you have to spend a lot of time and money researching its safety and efficacy,” says Jonathan Rousell, VP Generics Regulatory Affairs Europe at Teva.
“For a generic drug, the safety and efficacy for the intended use is already established. As such, it’s unnecessary and unethical to repeat those studies in full. As long as your product is the same in terms of the quantitative and qualitative properties of the drug substance, it meets the necessary quality standards and is bioequivalent, then you're not required to repeat all of those very expensive and very time-consuming studies that the brand originator did.
“Also, you're not building the brand, entering into marketing studies and there are fewer post-approval activities to confirm long term safety. In short, the product is the same in terms of its quality attributes and how it works in people, but you're not burdened with all those other things, so the development costs are much lower. That's where the price saving comes from.”
Securing regulatory approval
That’s not to say that developing and launching generic medicines is straightforward. Obtaining regulatory approval is a complex process, particularly at a company such as Teva which, in 2021, had more than 1,160 generic products in its development pipeline.
Maintaining a pipeline of generic drugs requires a lot of work behind the scenes − especially to gain regulatory approval from different regulators around the word. This is further complicated by the fact that regulations often differ by region and country and that we need to respect the intellectual property of the brand originator.
“It's quite complicated,” says Jonathan. “While we increasingly see alignment across the globe in the supporting data requirements, each country has its own regulator, whose remit is to protect public health. Precisely how they achieve that is a sovereign matter and therefore varies country to country.
“In the US you have the FDA [U.S Food and Drug Administration], one agency, working to one set of standards. Europe is a little more complex due to multiple countries and languages, but does have a common legal framework and decision-making process.
“Outside the US, EU and other highly regulated markets like Canada and Japan, many of the regulators in the rest of the world use something called regulatory reliance. This allows us to take our European or US dossiers largely as is and supplement them for local use. As the name suggests, those countries rely on the approval from another market, but depending on the country we may need to perform additional studies against the local brand originator. Overall, it’s a bit of a mixed bag.”
Globally, plans are afoot to implement international standards around the submission of data to regulatory authorities (so called “ISO IDMP”). This will further harmonize standards and ways of working, but will also greatly increase the amount and type of data to be submitted on generic drugs, Jonathan adds.
Working with regulators: the process
To increase the likelihood that Teva’s generic drugs are approved by regulators, a method called “Quality by Design” is employed in research and development. This method helps to maintain a high quality of medicines by using statistical, analytical and risk-management methodology in the design, development and manufacturing of medicines.
Another important task is the pre-approval phase - testing how well the drug’s dosage performs compared to the branded equivalent. After the generic drug product is developed, it goes through a submission and approval process, which can take a year or more.
One of the main hurdles for a generic medicine to clear is demonstrating “bioequivalence” with a branded drug. Bioequivalence is defined as how closely two or more different drugs with the same active ingredients produce the same clinical outcomes and whether the branded and generic medicine are absorbed by the patient’s body at a similar rate.
Once this has been established, the product receives its marketing authorization and, subject to legal regulation, launches on the market. Teva currently has 40,000 marketing authorizations across the world.
Teva’s medicines are taken by 200 million patients globally every day and we make a large number of the medicines listed by the World Health Organization as being the most effective and safe to meet the most important needs in a healthcare system. Around the world, 40,000 Teva employees in 55 sites help develop, produce and deliver the world’s biggest medicine cabinet.
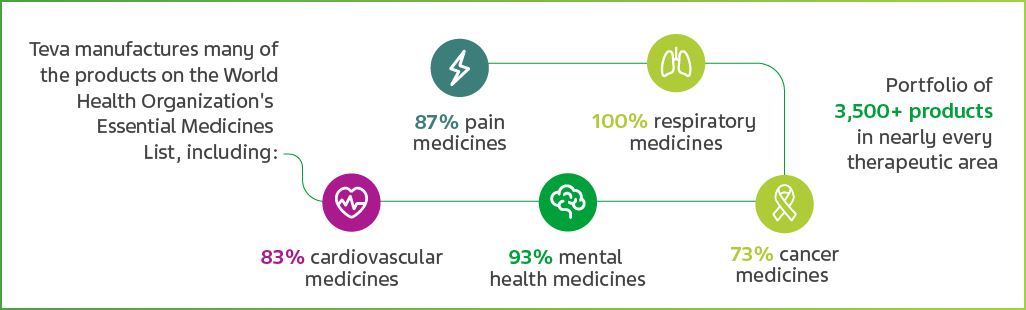
Teva’s contribution to world healthcare is affordable, high quality medicine. It is of vital importance to us that we continue to maintain that quality as we increase our reach and patients’ access to drugs. That’s our commitment.
Read more
- Producing generic medicines requires an extensive amount of research and development. Go behind the scenes at Teva to find out more
- Take a trip with us through the story of generic medicines – from what to why to who
- Want to be part of a team working to improve the lives of millions of people around the world? Visit our Careers section
You might also be interested












